



Next: Input file
Up: User's manual of ADPACK
Previous: Installing
Contents
If the installation is completed normally, move to the directory 'work',
and then you can perform the program, adpack, using an input file,
C.inp as follows:
% adpack C.inp
The test input file, C.inp, is for performing the SCF calculation of
a carbon atom. The calculation is performed in only several seconds
by a 2.4 GHz Xeon machine, although it is dependent on a computer.
When the calculation is completed normally, three files (C0.alog, C0.ao,
and C0.aden) are output to the directory, 'work'.
C0.alog is the log file of the calculation which includes the contents
of an input file, the convergence history in SCF steps,
and the total energy decomposed to the contributions.
A part of the file, C0.alog, is shown below. It is found that
the convergence is achieved by 12 SCF steps for the eigenvalues energy of
a Kohn-Sham equation, Eeigen, and the norm of the difference between
the input and output densities.
***************************************************
SCF history in all electrons calculations
***************************************************
SCF= 1 Eeigen=-31.1432610521402 (Hartree) NormRD= 9.7504824337909
SCF= 2 Eeigen=-31.2507824481920 (Hartree) NormRD= 9.6908568790503
SCF= 3 Eeigen=-29.2904374089900 (Hartree) NormRD= 6.4223342805654
SCF= 4 Eeigen=-24.3586103571626 (Hartree) NormRD= 1.3490158536346
SCF= 5 Eeigen=-21.9965036829842 (Hartree) NormRD= 0.1523028186916
SCF= 6 Eeigen=-21.5002109590127 (Hartree) NormRD= 0.0119067469939
SCF= 7 Eeigen=-21.3467192266812 (Hartree) NormRD= 0.0005718475963
SCF= 8 Eeigen=-21.3045977061498 (Hartree) NormRD= 0.0000175378857
SCF= 9 Eeigen=-21.2984619045622 (Hartree) NormRD= 0.0000005376916
SCF= 10 Eeigen=-21.2965170176425 (Hartree) NormRD= 0.0000000125540
SCF= 11 Eeigen=-21.2966277103150 (Hartree) NormRD= 0.0000000012975
SCF= 12 Eeigen=-21.2964361910017 (Hartree) NormRD= 0.0000000000864
The eigenvalues and the total energy, Etot, are also output in C0.alog.
***************************************************
Eigenvalues (Hartree) in all electrons calculations
***************************************************
n= 1 l= 0 -9.9479219357833
n= 2 l= 0 -0.5009865574917
n= 2 l= 1 -0.1993096022259
***************************************************
Energies (Hartree) in all electrons calculations
***************************************************
Eeigen = -21.2964361910017
Ekin = 37.1873926464442
EHart = 17.6249339614759
Exc = -4.7271002754349
Eec = -87.5097256776491
Etot = Ekin + EHart + Exc + Eec
Etot = -37.4244993451638
Figure:
(a) Electoron density of a carbon atom,
(b) Radial wave functions of a carbon atom
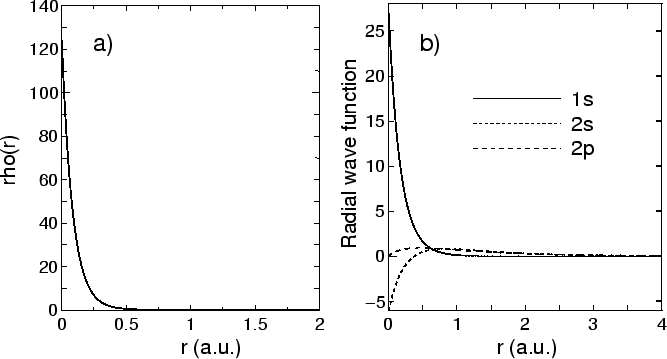 |
The electron density 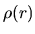 as a function of radius is output
in a file, C0.aden. Figure 1(a) shows electron density of a carbon atom
stored in C0.aden.
In the file, C0.aden, the first, second, third columns mean log(r),
r, and the electron density in all a.u., respectively.
The order of data is also similar in the other files.
The radial wave functions, shown in Fig. 1(b), are output in a file,
C0.ao, in which
they are listed in order of log (r), r, and the radial wave functions
of l=0 for n=1.
For n=2 or subsequent ones, radial wave functions are stored
in the same order as that for n=0.
However, note that the ingredients are output up to l=n-1 as follows:
as a function of radius is output
in a file, C0.aden. Figure 1(a) shows electron density of a carbon atom
stored in C0.aden.
In the file, C0.aden, the first, second, third columns mean log(r),
r, and the electron density in all a.u., respectively.
The order of data is also similar in the other files.
The radial wave functions, shown in Fig. 1(b), are output in a file,
C0.ao, in which
they are listed in order of log (r), r, and the radial wave functions
of l=0 for n=1.
For n=2 or subsequent ones, radial wave functions are stored
in the same order as that for n=0.
However, note that the ingredients are output up to l=n-1 as follows:
n=1
log(r), r, l=0
...............
n=2
log(r), r, l=0, l=1
....................
n=3
log(r), r, l=0, l=1, l=2
.........................
Next: Input file
Up: User's manual of ADPACK
Previous: Installing
Contents
2011-09-28