It is possible to optimize geometrical structures with a constraint
in which atoms can be fixed in the initial position.
The constraint can be applied separately to the 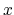 -,
-, 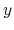 -, and
-, and  -coordinates
to the initial atomic position in your input file by
the following keyword 'MD.Fixed.XYZ':
-coordinates
to the initial atomic position in your input file by
the following keyword 'MD.Fixed.XYZ':
<MD.Fixed.XYZ
1 1 1 1
2 1 0 0
MD.Fixed.XYZ>
The example is for a system consisting of two atoms. If you have  atoms,
then you have to provide rows in this specification.
The 1st column is the same sequential number to specify atom as in the
specification of the keyword
'Atoms.SpeciesAndCoordinates'.
The 2nd, 3rd, and 4th columns are flags for the -, -, and -coordinates, respectively.
'1' means that the coordinate is fixed, and '0' relaxed.
In the above example, the -, -, and -coordinates of the atom '1' are fixed,
and only the x-coordinate of the atom '2' is fixed. The default setting is
that all the coordinates are relaxed. The fixing of atomic positions
are valid for all the geometry optimizers and molecular dynamics schemes.
The constrained relaxation may be useful for a refinement of the
local structure in large-scale systems.
atoms,
then you have to provide rows in this specification.
The 1st column is the same sequential number to specify atom as in the
specification of the keyword
'Atoms.SpeciesAndCoordinates'.
The 2nd, 3rd, and 4th columns are flags for the -, -, and -coordinates, respectively.
'1' means that the coordinate is fixed, and '0' relaxed.
In the above example, the -, -, and -coordinates of the atom '1' are fixed,
and only the x-coordinate of the atom '2' is fixed. The default setting is
that all the coordinates are relaxed. The fixing of atomic positions
are valid for all the geometry optimizers and molecular dynamics schemes.
The constrained relaxation may be useful for a refinement of the
local structure in large-scale systems.